In combined studies two different product types (medicinal product, medical device and/or in-vitro diagnostic) are being investigated. As a result, the study must be assessed on the basis of multiple EU-regulations, as shown in the table below:
| Possible combined studies | EU regulation |
|---|---|
| Medicinal product and medical device | CTR and MDR |
| Medicinal product and in vitro diagnostic | CTR and IVDR |
| Medicinal device and in vitro diagnostic | IVDR and MDR |
Within combined studies, the part of the study that falls under the CTR (Clinical Trial Regulation), IVDR (In-Vitro Diagnostics Regulation) and/or MDR (Medical Device Regulation) must be assessed by an authorized review committee. This often leads to many questions about combined studies. The most frequently asked questions can be found at the bottom of this page. Information on the specific rules and legal frameworks per EU-regulation can be found on the following pages:
Clinical trials with medicinal products (CTR)
Clinical investigations with medical devices (MDR)
Performance studies using in vitro diagnostics (IVDR)
Sponsors are strongly recommended to use our nationally coordinated process for the initial submission of a combined study. In this process you submit both research files simultaneously, allowing the validation and assessment phase to run in parallel. This process enables rapid decision-making and start of your combined study. More information on the coordinated process can be found below.
Introduction
The Netherlands offer sponsors a coordinated process for combined studies. In this process, the combined study is assessed simultaneously under both regulations. Within this coordinated process, a specific set of requirements applies, as described below and in the flowchart. Please note that within the coordinated process both studies remain legally independent, meaning that all relevant EU-regulations apply.
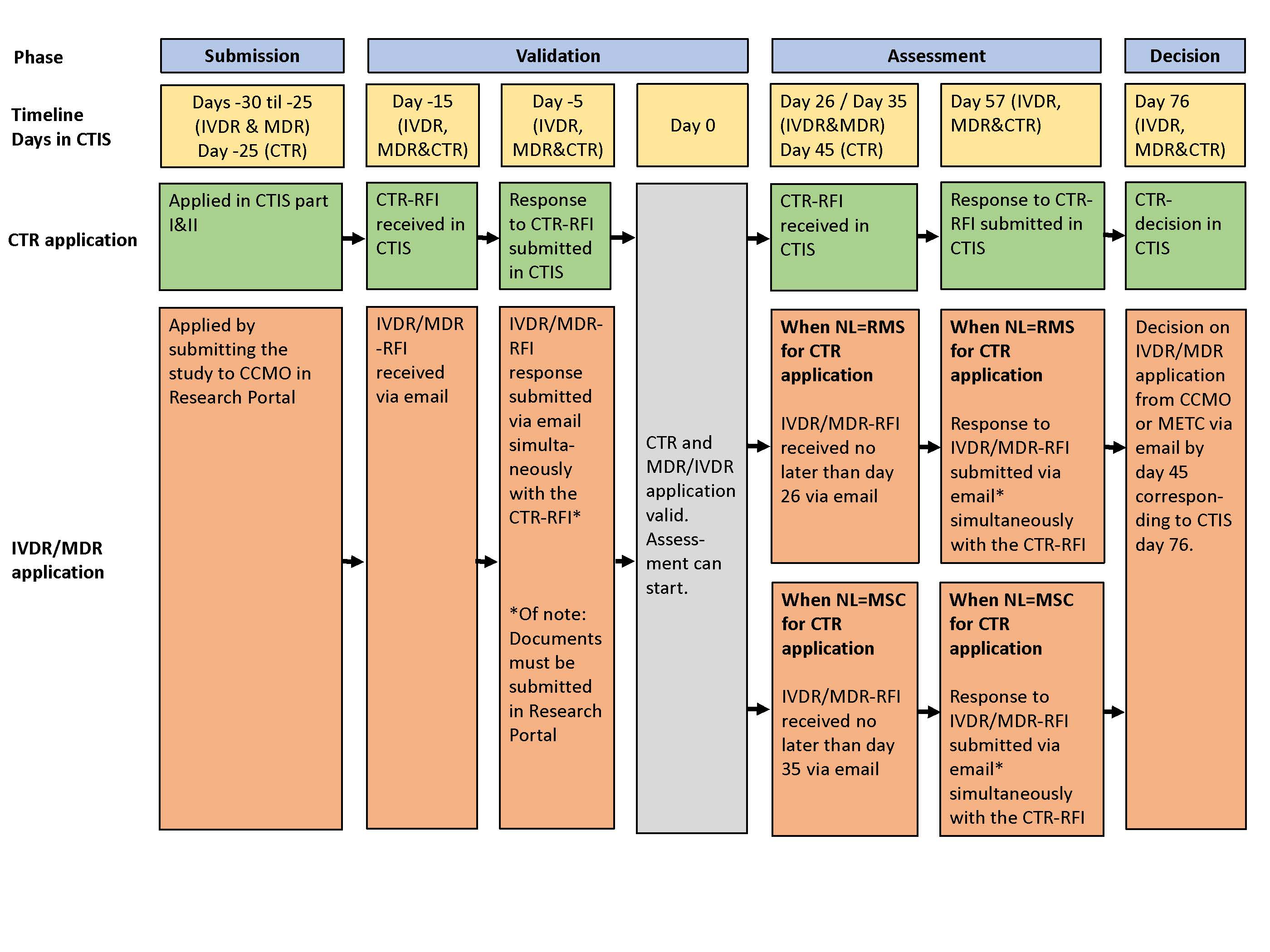
The coordinated process aims to harmonize the submission procedure of a CTR clinical trial (CTR study) with the submission procedure of an IVD performance study (IVDR study) or an MDR clinical investigation (MDR study). When the validation and assessment phase of the studies are performed in the Netherlands, they are coordinated by CCMO. The assessment under both regulations is carried out by the same authorized review committee and the decision always follows on day 76 according to the CTIS deadline. This coordinated process allows for a more rapid assessment, preventing a delay in the start of your combined study.
The coordinated process is shown in the flowchart below.
Image: © CCMO
Conditions
The following requirements apply if you want to use the coordinated process:
- The IVDR study and/or MDR study must be submitted earlier (up to 5 days) or at the same time as the CTR study;
- Validation and assessment of the IVDR study or MDR study are performed in parallel with the CTR study validation and assessment phase. The CTIS deadlines are applicable;
- Sponsors must submit the response to the RFI (request for information) for both studies simultaneously according to the timeline overview as shown in the flowchart;
- A decision is made for both studies on the same day (day 76 in CTIS). Only if the Netherlands is MSC the decision for the MDR/IVDR study will precede the CTR study;
- The coordinated process cannot be completed if the sponsor deviates from the timelines as shown in the flowchart;
- Parallel assessment and decision-making cannot be guaranteed if the CTR assessment is extended by 50 days or if consultation is requested based on MDR/IVDR with a period of 20 days.
Four phases coordinated process
The following requirements apply to the submission of a combined study:
- The CTR research file is submitted via CTIS. Part 1 and 2 of the research file must be submitted simultaneously.
- The MDR research file or IVDR research file is submitted via the Research Portal. This can be done up to 5 days before submission of the CTR study, but not after submission of the CTR study.
- Please send a notification to the CCMO via devices@ccmo.nl if you submitted an article 82 MDR study to a local METC. Please state the NL-number and which MREC has been selected.
- In the case of a combined study, two complete research files must be submitted, depending on the applicable EU-regulation. However, it may be possible to submit the same document. This may be applicable to the study protocol, research participant insurance, PIF (information letter and consent form) and VGO (site suitability declaration). When the same document is used, we request the following:
- Use the same version number of the document;
- Include both the title of the CTR research file and the IVDR/MDR research file in the document;
- Indicate in the cover letter of both research files which documents are the same.
When submitting the cover letter of both studies please include the following:
- Clearly indicate that your study is a combined study. Please state the CTIS- number and/or NL-number.
- Indicate your preferred MREC.
- Confirm that you are using the coordinated process and that the sponsor agrees to the proposed deadlines for responding to RFIs, following the CTIS deadlines.
- A list of documents that are the same in the CTR and the MDR or IVDR submissions (including version number and date).
The validation of both research files in a combined study is carried out in parallel. However, separate validation processes are followed. The validation will be coordinated in such a way that both studies are validated simultaneously, day 0 in CTIS. This means that if an RFI is only sent for one of the research files, an administrative RFI is sent for the other to keep the timelines parallel.
The assessment of a combined study is performed by the same authorized review committee for both regulations. The deadlines for sending and responding to RFI's are defined by the CTIS timelines as shown in the flowchart. Please note that separate procedures are followed for sending and responding to RFIs. There is a difference in the timelines, depending on the role that the Netherlands have in the CTR clinical trial (MSC or RMS). The exact timelines are shown in the flowchart.
In the coordinated process, the decision for both studies always follow shortly after each other. The applicable deadlines in CTIS are leading, as shown in the flowchart.
Frequently asked questions combined studies
Include the following points in the cover letter of both studies:
- Clearly indicate that it concerns a combined study. Please include the CTIS- number and/or NL-number.
- Indicate the preferred MREC.
- A list of documents that are the same for both submissions (including version number and date).
Combined studies are assessed based on multiple EU-regulations, each with specific rules. This means that they are legally two separate submissions. We strongly recommend that you use the nationally coordinated process for combined studies. This guarantees a rapid assessment and the start of your combined study. Please note that the flowchart only indicates combined studies under the CTR with MDR/IVDR. In case your study combines a medical device and an in-vitro diagnostic we also request to submit the studies simultaneously, allowing the validation and assessment phase to run in parallel.
{kind=link}
If you do not use the coordinated process, the following requirements still apply:
- In a combined study, two complete research files must always be submitted. You are not allowed to refer to the other research file.
- The CTR research file is submitted via CTIS.
- The MDR- or IVDR-research file is submitted via the Research Portal.
In a combined study, two complete research files must be submitted by law. The specific requirements for the content of the research files are described in the applicable EU-regulations. More information about the standard research file per EU-regulation can be found here:
Standard research file IVD performance studies
Some documents in the research file can be the same for both the CTR research file and the MDR/IVDR research file. This includes, for example, the research protocol, research participant insurance, SIS (information letter and consent form) and VGO (site suitability declaration). If the same document is used, we request the following:
- Use the same version number;
- Include both the title of the CTR file and that of the MDR/IVDR file in the document;
- State both the NL and CTR numbers explicitly in the site suitability declaration (VGO).
- Indicate in the cover letter of both research files which documents are the same;
- In case a single protocol is used it must contain primary objectives and endpoints for both studies.
The following principle can be used to make a decision between a single SIS or two SIS's in a combined study:
- In most cases, one SIS will be desirable. If the studies are integrated in such a way (e.g. in studies with a companion diagnostic) that it can be seen as a single study, it is desirable to present one SIS to the research participant. Informed consent can then be given to both studies with one signature. Of note, it must be very clear to the research participant that consent is given for both studies. This can be done, for example, by having the research participant tick a box for both studies.
- In a number of cases, two SIS's will be desirable. This is the case when only a small percentage of research participants from the IVD performance study are suitable for participation in the CTR clinical trial. In this case, informed consent must also be given separately.
If you do not make use of the coordinated process, the start of your combined study may be delayed.
It is possible to submit a so-called ‘umbrella’ IVDR study that is linked to multiple CTR studies.
An amendment for a combined study can apply to one study or to both studies. If the amendment results in a change to a shared document, the amendment must be submitted for both studies. The sponsor decides whether it is a substantial amendment for both studies or only a notification for one or both studies. It is up to the review committee to determine whether this decision is correct. If the amendment is substantial for both studies, we advise you to submit the amendment at the same time. If the amendment is substantial for only one study, we advise you to submit the substantial amendment first. After approval you can submit the notification to the other study.
Amendments for combined studies are submitted separately. This means that an amendment under the CTR is submitted in CTIS and amendments under the MDR/IVDR in the Research Portal. More information on submitting amendments per EU regulation can be found here:
Modifications (CTR)
Modifications of the research file (amendments) (MDR)
Modifications of the research file (amendments) (IVDR)
Different legal requirements apply to safety reports. For example, something may have to be reported under the CTR and not under the MDR/IVDR. The safety report for combined research must be submitted separately.
For combined studies, the specific appeal and objection procedures continue to apply to the part of the study that falls under the IVDR, MDR or CTR. Information about starting an appeal or objection procedure against a decision of the authorized MREC or the CCMO can be found on the following pages:
After approval of your combined study, you must submit data during the study, upon completion and after completion of the study . The data you must submit for a combined study depends on the legal framework that applies to the combined study. No additional requirements are imposed for combined studies. Information about this per review framework can be found on the following pages:
During and after IVD performance studies