Investigational and auxiliary medicinal products (i.e. IMPs and AxMPs), regardless of their legal status, should be appropriately labelled in order to ensure the safety of the clinical trial participants and the reliability and robustness of data generated in clinical trials, and to allow for the distribution of those products to clinical trial sites throughout the EU.
Labelling information should be included in the application dossier, specifically in the clinical trial application in CTIS, under the category "Content labelling of the IMPs". A mock-up of the label is not required, only the textual content needs to be submitted.
Labelling requirements for investigational medicinal product and auxiliary medicinal products are laid down in Chapter X and Annex VI of the CTR and further elaborated in the CTR Q&A.
Please refer to the labelling decision tree and to the subsequent sections for the general and specific labelling requirements for the different categories of investigational and auxiliary medicinal products.
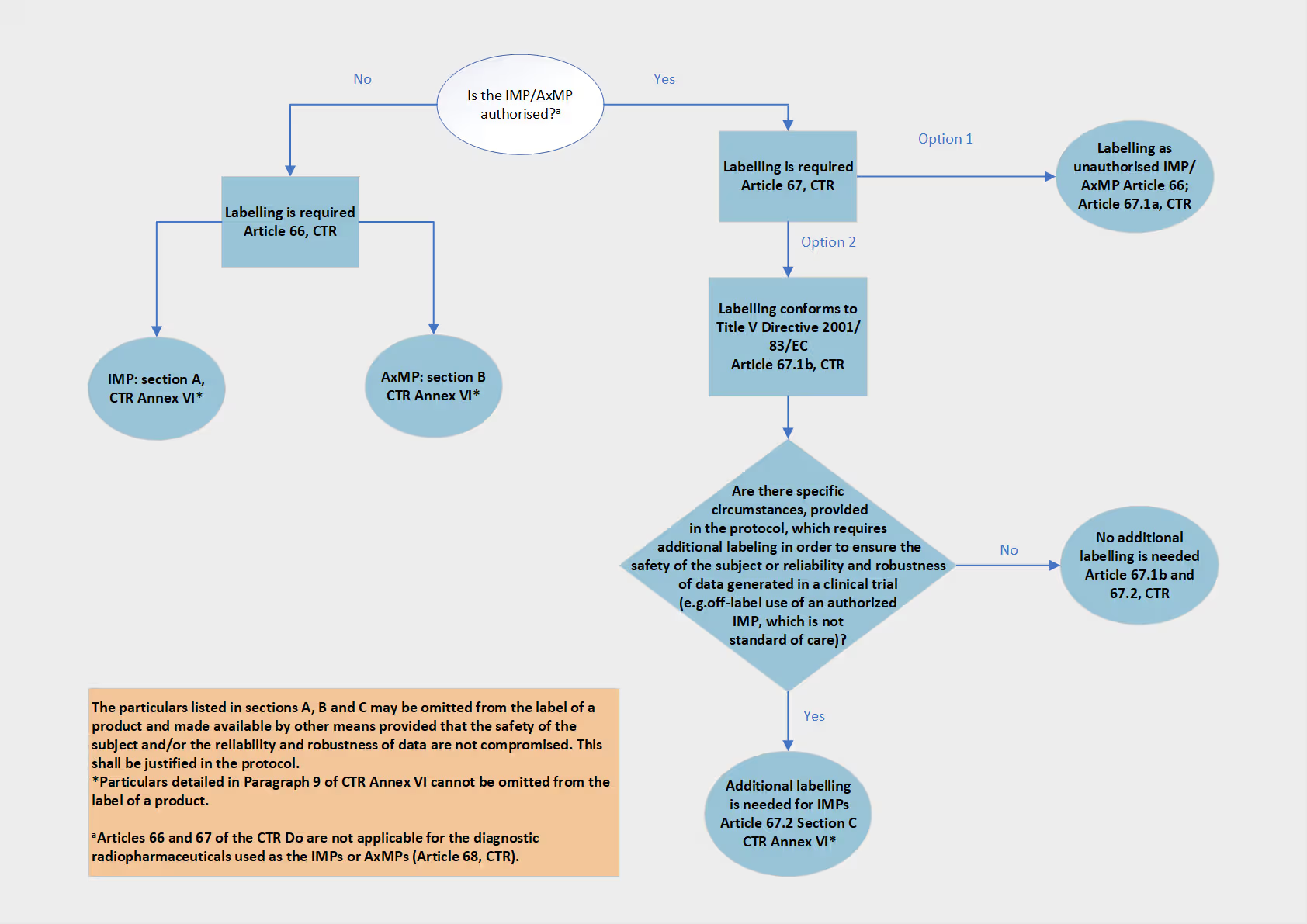
General notes for labelling content on IMPs/ AxMPs to be used in clinical trials
Symbols or pictograms may be used to further clarify the labelling information. However, symbols are only allowed for clarification and not to replace the mandatory text (refer to Annex VI Section A.1 paragraph 2). Additional information, warnings or handling instructions may be included.
In line with Annex VI Section A.1 paragraph 3 (applicable to unauthorised IMPs and AxMPs) the address and telephone number of the main trial contact are not required on the label if this information is presented by other means. Therefore, if the sponsor wishes to omit the address and telephone number of the main trial contact from the label the sponsor should confirm that the main trial contact information is available to the trial participants at all times on a patient card or any other document intended for the trial participants. Confirmation of the availability of the main trial contact information (for emergency purposes) should be provided in the cover letter (see "Key Document List" of the CTCG).
Of note:
- the name of the main trial contact is still required on the label (for authorised IMPs and AxMPs this is only required when an additional label is needed);
- only the contact information may be omitted (applicable to unauthorised IMPs and AxMPs);
- the name of the investigator should also be added to the label should this not be the same as the main trial contact.
In Annex II of the CTR Q&A the language requirements for the labels are detailed for each Member State. The language of the text on the label should be Dutch for studies performed in the Netherlands. However, English language text may also be acceptable in certain situations, e.g. if the investigational medicinal product is only administered by a qualified healthcare professional and is not handed out to the patient for use at-home. Using multiple languages (in addition to Dutch) on the same label is allowed (e.g. booklet label).
Labelling activities should generally be performed in accordance with the GMP requirements. For additional information refer to: Documentation relating to compliance with Good Manufacturing Practice (GMP) for Investigational Medicinal Product and Auxiliary Medicinal Product on the CCMO website.
Further details regarding the particulars required on the label for unauthorised investigational and auxiliary medicinal products are outlined in CTR Chapter X and further specified in (Sections A, B, and D).
Some information may be omitted from the label of an unauthorised medicinal product and made available by other means, provided that the safety of the trial participants and the reliability and robustness of data are not compromised (refer to Annex VI, Section D.8).
If the sponsor wishes to omit information (other than listed in Annex VI, Section D.9) and make this information available in another way this shall be adequately justified in the protocol. An example is the use of a centralised electronic randomisation system or centralised information system.
It is of importance to note that some specific information may not be omitted from the label (see Annex VI, D, paragraph 9a – d) and cannot be made available by other means. On a case-by-case basis, adequacy of particulates on the labelling content information will be assessed to assure the safety of the clinical trial participants and the reliability and robustness of the data obtained in the clinical trial.
Period-of-use information on the immediate and outer packaging of unauthorised medicinal products used in clinical trials may be regularly updated based on stability data. However, in certain cases frequent re-labelling of the immediate packaging may be associated with potential risks affecting the quality and safety of those products. In certain situations, such as when the immediate and outer packaging are provided together, or when the immediate packaging is in the form of blister packs or small units (small immediate packaging), it is appropriate to omit the period of use from the immediate packaging. However, the information on the period of use and any other potentially omitted information on the immediate packaging still needs to be reported on the outer packaging as outlined in Annex VI, paragraph 1 (a reference is made to Annex VI, paragraph 4 and 5).
Certain unauthorised medicinal products require reconstitution/dilution prior to administration to patients. In such cases, labelling in line with the requirements outlined in Annex VI are applicable to the original primary and secondary packaging. However, the label that will be prepared after reconstitution/dilution of the unauthorised medicinal product does not need to be submitted for evaluation; the pharmacist is responsible for the content of the latter label, which should still conform to the national requirements (NVZA).
As a general rule no additional labelling is required for investigational and auxiliary medicinal products which are subject to a marketing authorisation. More specifically, they may be labelled as specified in Title V of Directive 2001/83/EC, where no additional information is required. Alternatively, authorised IMPs and AxMPs may also be labelled in line with the requirements outlined in Chapter X, Article 66, i.e. in line with the requirements for unauthorised investigational and auxiliary medicinal products. Please refer to the section "Labelling for unauthorised IMPs and AxMPs" above for details.
When authorised IMPs and AxMPs are labelled in accordance with Title V of Directive 2001/83/EC, and the specific circumstances require so in order to ensure the safety of the research participant or the reliability and robustness of data generated in the clinical trial, additional labelling information relating to the identification of the clinical trial and of the contact person on the outer and immediate packaging may be necessary (refer to Chapter X, Article 67, Paragraph 2). The specific circumstances should be outlined in the protocol. A list of the additional particulars required on the outer and immediate packaging is outlined in Section C of Annex VI.
The following examples of clinical trials with authorised investigational medicinal products may require additional content labelling information or a full label (as outlined in Chapter X, Article 67, Paragraph 2):
- Over-encapsulation or other means of modification are used, for instance, to ensure blinding of an authorized investigational or auxiliary medicinal product. Modifications may also be performed to obtain a different strength than the original authorised medicinal product. Please note that in such instances the authorized medicinal product is regarded as modified authorised and as such should be fully labelled in accordance with labelling requirements of unauthorised medicinal products. Please refer to section ‘Labelling of unauthorised IMPs and AxMPs’ for further guidance.
- An authorised medicinal product is used for the same medical condition as laid down in the corresponding SmPC, but this form of treatment is not considered the standard of care in the trial's target population. For instance, a clinical trial with minors where the IMP is authorised in adults only and the use in (this age group of) children is not part of the current paediatricians’ treatment guidelines.
- An authorised medicinal product is used outside of the authorised indication (“off-label use”), but this form of treatment is standard of care in the target population. However, as the dosing scheme or route of administration deviates from that laid down in the patient information leaflet, an additional label may need to be provided.
- An authorised medicinal product is provided with an EU/EEA commercial label, but not in Dutch/English language (see paragraph “General notes for labelling content on IMPs/ AxMPs to be used in clinical trials” for details) while the clinical trial is being conducted in the Netherlands. An additional label or complete re-labelling in Dutch may be required. However, in this case the translated label does not need to be submitted for assessment.
In summary, a risk-based approach should be followed regarding the need for additional labelling in case of use outside the authorised indication of an authorised investigational medicinal product. A justification for either omission of an additional label or for the need for an additional label should be laid down in the protocol in case of use outside the authorised indication.
The sponsor should consider the authorised medical condition, the authorised patient group, the authorised route of administration, and any other relevant particulates laid out in the SmPC when deciding whether additional labelling information is needed for an authorised medicinal product or whether it could be omitted altogether. Regardless of sponsor-identified risks, the CCMO/MRECs may request additional labelling if deemed necessary for trial participant safety and/ or data reliability.
As with non-authorised products, certain information may also be omitted from the label of an authorised medicinal product and made available in another way, provided that the safety of the trial participants and the reliability and robustness of data are not compromised. If the sponsor wishes so, adequate justification should be submitted (refer to Section D of Annex VI).
Labelling requirements conforming to CTR are not applicable for radiopharmaceuticals used as diagnostic investigational or as diagnostic auxiliary medicinal products (as outlined in general provision [57] and Article 68). However, these products should still be labelled appropriately to ensure the safety of the clinical trial participants and the reliability and robustness of data generated in the clinical trial.
Generally, the sponsor is expected to adhere to the particulates outlined in the Annex VI, sections A, B, and D, as well as for the labelling of diagnostic radiopharmaceuticals. Please refer to the section "Labelling for unauthorised IMPs and AxMPs" above for details on labelling requirements. In addition, due to the nature of these products, the radioactivity of the IMP or AxMP needs to be listed in international units (Becquerel [Bq]), and a pictogram depicting radioactivity should be denoted on the content labelling information as well.
Magistral and officinal preparations used as investigational and auxiliary medicinal products should be labelled according to the CTR requirements for unauthorised investigational and auxiliary medicinal products. Please refer to the section "Labelling for unauthorised IMPs and AxMPs" above for details on labelling requirements. However, should magistral and officinal preparations be used as auxiliary medicinal products, the content labelling information does not need to be submitted.